
Topic 2: Targeted and Non-Targeted Toxicity
LEARNING OBJECTIVES
After completing this lesson, you will be able to:
After completing this lesson, you will be able to:
- Discuss the differences between targeted and non-targeted toxicity.
- Explain how absorption, distribution, and metabolism of a toxicant might influence its effects in the body.
- Discuss types of interactions that can occur between a toxicant and its target molecule and how they impact the host.
- Explain the importance of bioactivation in the pathophysiology of acetaminophen toxicosis.
Toxic effects occur when the ultimate toxicant reacts with a host molecule, cell or tissue, triggering a secondary series of events leading to toxic injury. The ultimate toxicant may be the original xenobiotic to which the host was exposed, a metabolite of the original xenobiotic, a reactive species generated by the original xenobiotic or its metabolite, or an altered or unchanged endogenous compound. Some toxicants can interact with any compound they encounter, while other toxicants exert their effects by interacting with specific sites (targets) on molecules. The types of interaction between toxicants and their targets include:
Covalent binding forms practically irreversible bonds between the toxicant and target molecule; the resulting toxicant-target molecule is termed an adduct. Covalent binding occurs most commonly when electrophilic (electron-seeking) compounds join with nucleophilic (electron-donating) compounds. Nucleophilic compounds are abundant in biological systems, and include cellular macromolecules such as proteins, nucleic acids, and phospholipids.
Covalent binding of toxicants to cellular proteins can inhibit vital enzyme reactions, alter protein function, inhibit or stimulate membrane receptors, or damage membrane proteins. Covalent binding of toxicants to nucleic acids such as RNA and DNA can inhibit or alter protein synthesis or induce DNA mutations that can lead to cellular dysfunction or carcinogenesis. Covalent binding of toxicants to phospholipids can result in direct damage to cellular or organelle membranes or can trigger lipid peroxidation, which will be discussed later.
- Covalent binding
- Noncovalent binding
- Electron transfer
- Hydrogen abstraction
- Enzymatic reactions
Covalent binding forms practically irreversible bonds between the toxicant and target molecule; the resulting toxicant-target molecule is termed an adduct. Covalent binding occurs most commonly when electrophilic (electron-seeking) compounds join with nucleophilic (electron-donating) compounds. Nucleophilic compounds are abundant in biological systems, and include cellular macromolecules such as proteins, nucleic acids, and phospholipids.
Covalent binding of toxicants to cellular proteins can inhibit vital enzyme reactions, alter protein function, inhibit or stimulate membrane receptors, or damage membrane proteins. Covalent binding of toxicants to nucleic acids such as RNA and DNA can inhibit or alter protein synthesis or induce DNA mutations that can lead to cellular dysfunction or carcinogenesis. Covalent binding of toxicants to phospholipids can result in direct damage to cellular or organelle membranes or can trigger lipid peroxidation, which will be discussed later.
DID YOU KNOW?
Exposure to the analgesic drug acetaminophen is the most common cause of drug-induced liver injury in humans, accounting for 30,000 hospitalizations and approximately 1200 liver transplants performed every year in the United States. Most cases of acetaminophen-induced liver injury are the result of accidental or intentional overdoses. However, certain risk factors can increase the chance of liver injury developing at therapeutic doses of acetaminophen. These risk factors include chronic alcohol consumption, ingestion of drugs or herbs that alter liver antioxidant levels or pathways, malnourishment or advancing age with subsequent impairment or decline of normal liver metabolic pathways, and presence of chronic liver disease (e.g. viral hepatitis).
Exposure to the analgesic drug acetaminophen is the most common cause of drug-induced liver injury in humans, accounting for 30,000 hospitalizations and approximately 1200 liver transplants performed every year in the United States. Most cases of acetaminophen-induced liver injury are the result of accidental or intentional overdoses. However, certain risk factors can increase the chance of liver injury developing at therapeutic doses of acetaminophen. These risk factors include chronic alcohol consumption, ingestion of drugs or herbs that alter liver antioxidant levels or pathways, malnourishment or advancing age with subsequent impairment or decline of normal liver metabolic pathways, and presence of chronic liver disease (e.g. viral hepatitis).
Figure 2.1 Liver transplantation surgery. Acetaminophen toxicosis is the leading cause of drug-induced liver injury leading to transplantation.
Acetaminophen itself does not itself cause liver injury. Under normal circumstances, acetaminophen is converted in the liver primarily into non-toxic metabolites that are then excreted from the body. However, about 10% of acetaminophen is bioactivated by CYP450 enzymes to a toxic metabolite called N-acetyl-p-benzoquinoneimine (NAPQI); under normal circumstances, the liver can neutralize NAPQI by binding it to the antioxidant molecule glutathione. However, in overdose situations or when glutathione levels are low (as in chronic liver disease or malnutrition), the amount of NAPQI formed exceeds the capacity of glutathione to bind it, and instead NAPQI covalently binds onto macromolecules in the hepatocyte, causing hepatocellular injury and death. Treatment of acetaminophen overdose frequently involves administering antioxidants and glutathione precursors, such as N-acetylcysteine, in an attempt to provide alternative molecules to which NAPQI may bind.
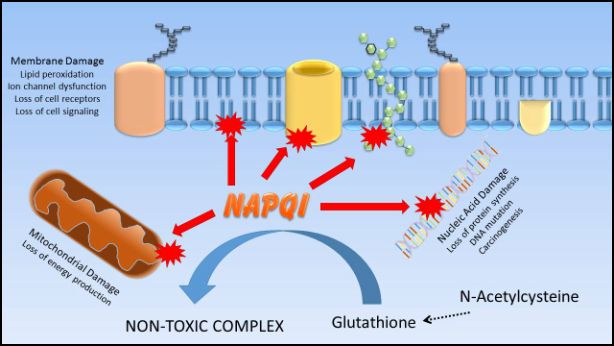
Figure 2.2 The acetaminophen metabolite NAPQI is normally detoxified through binding with glutathione. If glutathione becomes depleted, NAPQI can then bind to cellular macromolecules, resulting in cell injury. Providing the glutathione precursor n-acetylcysteine helps to replenish glutathione stores and mitigate NAPQI-induced injury to cellular structures.
Noncovalent binding of toxicants to target molecules involves the formation of relatively weak hydrogen or ionic bonds. Because of the relatively weak bonds formed, noncovalent binding is usually reversible. Noncovalent binding is commonly seen with toxicants that interact with cellular membrane receptors, intracellular receptors, some enzyme systems and membrane ion channels. Noncovalent bonding to these structures may result in over- or under stimulation of normal function. These interactions can result in alteration of inter- and intracellular signalling, organelle dysfunction, alteration of enzyme function, interference with normal homeostatic ion movement, general cellular dysfunction, or cell death.
Electron transfer can result in oxidation of some endogenous macromolecules, altering their function. For example, chemicals that oxidize ferrous iron (Fe++) to ferric iron (Fe+++) in the hemoglobin molecule cause a condition termed methemoglobinemia, resulting in lowered ability of the hemoglobin in red blood cells to deliver oxygen to body tissues. Electron transfers can result in the formation of radicals, highly reactive atoms or molecules with unpaired electrons that can bind to and damage cellular molecules and structures. Oxygen-derived free radicals ("free" because they are not attached to larger molecules) such as superoxide ion (•O2-) and hydroxyl radical (HO•) are produced during normal physiological processes, but when present in excess can cause significant cellular injury due to their ability to self-perpetuate. Antioxidants present in living organisms help mitigate this “snowball effect” by donating electrons without forming radical species. Important antioxidants present within cells include superoxide dismutase, catalase, vitamin A, and glutathione.
Electron transfer can result in oxidation of some endogenous macromolecules, altering their function. For example, chemicals that oxidize ferrous iron (Fe++) to ferric iron (Fe+++) in the hemoglobin molecule cause a condition termed methemoglobinemia, resulting in lowered ability of the hemoglobin in red blood cells to deliver oxygen to body tissues. Electron transfers can result in the formation of radicals, highly reactive atoms or molecules with unpaired electrons that can bind to and damage cellular molecules and structures. Oxygen-derived free radicals ("free" because they are not attached to larger molecules) such as superoxide ion (•O2-) and hydroxyl radical (HO•) are produced during normal physiological processes, but when present in excess can cause significant cellular injury due to their ability to self-perpetuate. Antioxidants present in living organisms help mitigate this “snowball effect” by donating electrons without forming radical species. Important antioxidants present within cells include superoxide dismutase, catalase, vitamin A, and glutathione.
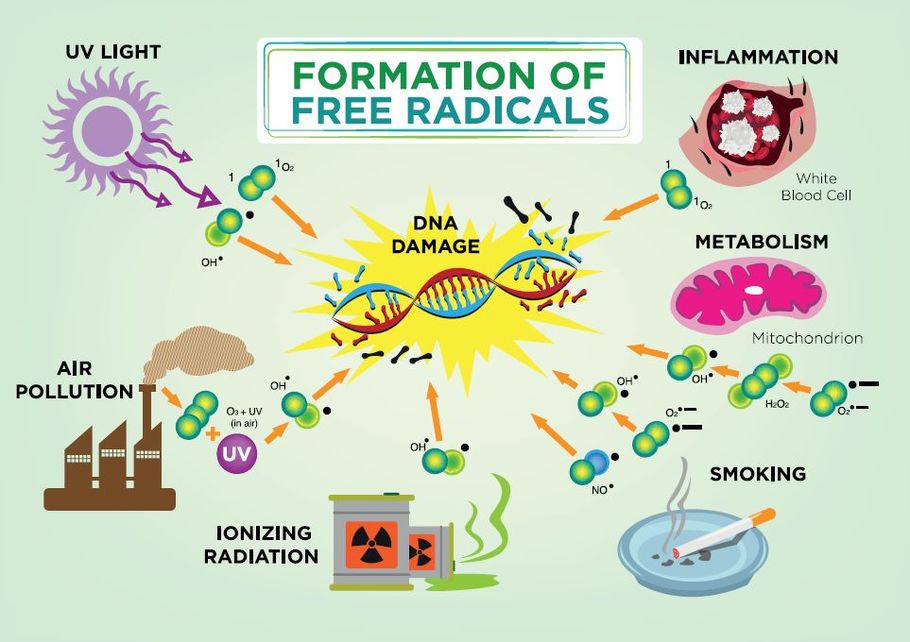
Figure 2.3 The formation and effects of free radicals.
Toxicant-induced hydrogen extraction from membrane phospholipids can induce lipid peroxidation which results in membrane injury leading to leakage and/or cell death. Lipid peroxidation occurs when an electrophilic compound steals an electron from a membrane phospholipid, producing a fatty acid radical. This fatty acid radical is relatively unstable and will react readily with molecular oxygen to produce a peroxyl-fatty acid radical. The peroxyl fatty acid radical then reacts with another fatty acid, producing yet another lipid peroxide and fatty acid radical that continues the cycle in a chain reaction-type mechanism. When present in sufficient quantities, antioxidants such as vitamin E can help terminate lipid peroxidation.
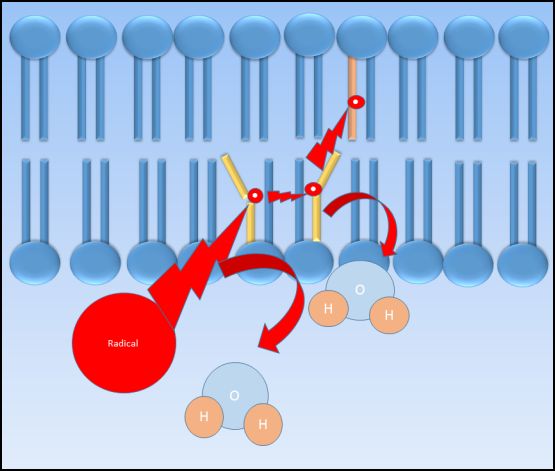
Figure 2.4 Cartoon of lipid peroxidation. An electrophilic radical interacts with the fatty acid chain of the cell membrane, extracting a hydrogen atom and producing a fatty acid radical which in turn interacts with other cell membrane fatty acids, propagating the production of radicals within the membrane. Lipid peroxidation may result in severe membrane injury if not terminated through interaction with an antioxidant.
Some toxicants act enzymatically with target molecules to exert their adverse effects. Ricin, a toxin from the castor bean plant, enzymatically hydrolyzes a bond in the ribosomal RNA molecule, thus blocking protein synthesis. Many snake venoms contain a variety of enzymes that can destroy cells and tissues of the body; for example, hyaluronidase cleaves connective tissue via hydrolysis of hyaluronic acid, an essential polysaccharide in connective tissue.

Figure 2.5 Rattlesnake venom contains a variety of enzymes and enzyme co-factors that cause tissue destruction
Non-Targeted Toxicity
Not all toxic effects are mediated through direct action of toxicant at the site of injury. Some toxicants exert their effects by altering the biological microenvironment of the affected cells or tissues, and the resulting injury may be distant from the site of toxicant action. For instance toxic ergot alkaloids produced by the fungus Claviceps purpurea act on blood vessels to cause vasoconstriction, resulting in reduced blood circulation and gangrene of the extremities. Other toxicants may exert their effects merely by being physically present, as when inert gases such as methane displace oxygen in the environment, resulting in asphyxia. Some xenobiotics can precipitate as crystals in the kidney tubules, resulting in mechanical damage to the tubular epithelium, leading to kidney injury.
Not all toxic effects are mediated through direct action of toxicant at the site of injury. Some toxicants exert their effects by altering the biological microenvironment of the affected cells or tissues, and the resulting injury may be distant from the site of toxicant action. For instance toxic ergot alkaloids produced by the fungus Claviceps purpurea act on blood vessels to cause vasoconstriction, resulting in reduced blood circulation and gangrene of the extremities. Other toxicants may exert their effects merely by being physically present, as when inert gases such as methane displace oxygen in the environment, resulting in asphyxia. Some xenobiotics can precipitate as crystals in the kidney tubules, resulting in mechanical damage to the tubular epithelium, leading to kidney injury.
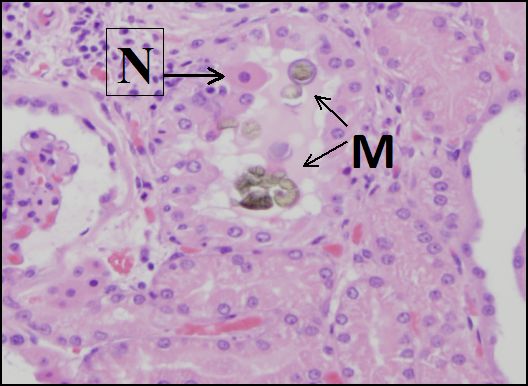
Figure 2.6 Precipitation of melamine-cyanuric acid crystals (M) causes mechanical damage to cells in the distal tubules of a canine kidney. A necrotic (dead) cell (N) has detached from the basement membrane. Melamine contamination of pet food caused kidney injury to hundreds of dogs and cats in 2007. (Courtesy of University of California, Davis Anatomic Pathology.)
Topic 2: Key Points
In this section, we explored the following main points:
In this section, we explored the following main points:
- Toxicants may interact with host molecules in a targeted or non-targeted fashion.
- Targeted interactions include covalent and noncovalent binding, electron transfer, hydrogen abstraction and enzymatic reactions.
- Covalent bonding of toxicant and target molecules form adducts, which can cause physical or functional damage to macromolecules such as proteins and nucleic acids.
- The covalent binding of the acetaminophen metabolite, NAPQI, to macromolecules in hepatocytes can result in severe liver injury requiring liver transplantation.
- Noncovalent binding between toxicant and target molecules form relatively weak, reversible bonds.
- Electron transfer can result in formation of self-perpetuating radicals that can cause significant damage to cellular structures.
- Antioxidants such as superoxide dismutase bind free radicals and help prevent perpetuation of free radical formation.
- Lipid peroxidation begins with extraction of hydrogen from membrane phospholipids, resulting in fatty acid radical formation and damage to membranes.
- Toxicants can act as enzymes, damaging cellular or tissue structures or interfering with normal cellular function.
- Non-targeted interactions include alterations in biological microenvironment of cell and tissues and impairment of normal function by physical obstruction or mechanical damage.


|

|
|
|